Fuel Property Prediction Model
FuelLib utilizes the group contribution method (GCM), as developed by Constantinou and Gani[1] [2] in the mid-1990s, to provide a systematic approach for estimating the thermodynamic properties of pure organic compounds. The GCM decomposes molecules into structural groups, each contributing to a target property based on predefined group values. By summing these contributions, the GCM accurately predicts essential properties, including the acentric factor, normal boiling point, liquid molar volume at standard conditions (298 K) and more. This predictive capability is particularly useful for complex mixtures such as synthetic aviation turbine fuels (SATFs), where experimental thermodynamic data is limited. FuelLib provides SATF developers with a means to estimate these critical properties without extensive physical testing, thereby aiding in the identification of promising fuel compositions before committing to large-scale production.
FuelLib builds on Pavan B. Govindaraju’s Matlab implementation, and includes gas chromatography data (GC x GC) for various jet fuels from the National Jet Fuel Combustion Program[3] (NJFCP) Air Force Research Laboratory[4] [5] (AFRL) and Vozka et al.[6]. Additionally, FuelLib includes correlations for the thermodynamic properties of mixture such as density, viscosity, vapor pressure, surface tension, and thermal conductivity. Table of GCM properties outlines the properties for the i-th compound in a mixture, which depends on the k-th first-order and j-th second-order group contributions.
Table of GCM properties
Property |
Units |
Group Contributions |
Units |
Description |
|---|---|---|---|---|
\(M_{w,i}\) |
kg/mol |
\(m_{w1k}\) |
g/mol |
Molecular weight. |
\(T_{c,i}\) |
K |
\(t_{c1k}\), \(t_{c2j}\) |
1 |
Critical temperature[1]. |
\(p_{c,i}\) |
Pa |
\(p_{c1k}\), \(p_{c2j}\) |
bar-0.5 |
Critical pressure[1]. |
\(V_{c,i}\) |
m3/mol |
\(v_{c1k}\), \(v_{c2j}\) |
m3/kmol |
Critical volume[1]. |
\(T_{b,i}\) |
K |
\(t_{b1k}\), \(t_{b2j}\) |
1 |
Normal boiling point[1]. |
\(T_{m,i}\) |
K |
\(t_{m1k}\), \(t_{m2j}\) |
1 |
Normal melting point[1]. |
\(\Delta H_{f,i}\) |
J/mol |
\(h_{f1k}\), \(h_{f2j}\) |
kJ/mol |
Enthalpy of formation at 298 K[1]. |
\(\Delta G_{f,i}\) |
J/mol |
\(g_{f1k}\), \(g_{f2j}\) |
kJ/mol |
Standard Gibbs free energy at 298 K[1]. |
\(\Delta H_{v,\textit{stp},i}\) |
J/mol |
\(h_{v1k}\), \(h_{v2j}\) |
kJ/mol |
Enthalpy of vaporization at 298 K[1]. |
\(\omega_i\) |
1 |
\(\omega_{1k}\), \(\omega_{2j}\) |
1 |
Acentric factor[2]. |
\(V_{m,\textit{stp},i}\) |
m3/mol |
\(v_{m1k}\), \(v_{m2j}\) |
m3/kmol |
Liquid molar volume at 298 K[2]. |
\(C_{p,i}\) |
J/mol/K |
\(C_{pA1_k}\), \(C_{pA2_k}\),… |
J/mol/K |
Equations for GCM properties
The properties of each compound in a mixture can be calculated as the sum of contributions from the first- and second-order groups that make up the compound. For a given mixture, let \(\mathbf{N}\) be an \(N_c \times N_{g_1}\) matrix that represents the number of first-order groups in each compound, where \(N_c\) is the number of compounds in the mixture and \(N_{g_1}\) is the total number of first-order groups as defined by Constantinou and Gani[1][2]. Similarly, let \(\mathbf{M}\) be an \(N_c \times N_{g_2}\) matrix that specifies the number of second-order groups in each compound, where \(N_{g_2}\) is the total number of second-order groups. The total number of groups \(N_g = N_{g_1} + N_{g_2} = 121\). The GCM properties for the i-th compound in the mixture are calculated as follows[1] [2] [8]:
Equations for individual compound correlations
This section presents correlations for physical properties that leverage the individual compound properties defined in Equations for GCM properties. These correlations make it possible to evaluate physical properties at non-standard temperatures and pressures, given that group contribution properties are only defined at standard conditions. Unless noted otherwise in the individual correlation, all units are assumed to be SI: length (m), mass (kg), time (s), temperature (K), mole (mol). The Reduced temperature quantities are used throughout this section for each compound i, provided \(T\) in K unless noted otherwise.
Property |
Units |
Description |
|---|---|---|
\(\nu_i\) |
m2/s |
Kinematic viscosity[9]. |
\(L_{v,\textit{stp},i}\) |
J/kg |
Latent heat of vaporization at 298 K[10]. |
\(L_{v,i}\) |
J/kg |
Temperature-adjusted latent heat of vaporization at 298 K[10]. |
\(V_{m,i}\) |
m3/mol |
|
\(\rho_i\) |
kg/m3 |
Density |
\(C_{\ell,i}\) |
J/kg/K |
Liquid specific heat capacity[10]. |
\(p_{sat,i}\) |
Pa |
|
\(\sigma_i\) |
N/m |
Surface tension[15]. |
\(\lambda_i\) |
W/m/K |
Thermal conductivity[8]. |
Symbol |
Definition |
Description |
|---|---|---|
\(T_{r,i}\) |
\(\frac{T}{T_{c,i}}\) |
Reduced temperature. |
\(T_{r,b,i}\) |
\(\frac{T_{b,i}}{T_{c,i}}\) |
Reduced boiling point temperature. |
\(T_{r,\textit{stp},i}\) |
\(\frac{298 \text{ (K)}}{T_{c,i}}\) |
Reduced standard temperature. |
Kinematic viscosity
- fuel.viscosity_kinematic(T, comp_idx=None)
Calculate the viscosity using Dutt’s equation.
- Parameters:
T (float) – Temperature in Kelvin.
comp_idx (int, optional) – Index of compound to calculate property for.
- Returns:
Viscosity of each component in m^2/s.
- Return type:
np.ndarray
The kinematic viscosity of the i-th compound of the fuel,
is calculated from Dutt’s equation (Eq. 4.23 in Viscosity of Liquids[9]) provided \(T\) in \(^{\circ}\) C:
Latent heat of vaporization
- fuel.latent_heat_vaporization(T, comp_idx=None)
Calculate latent heat of vaporization adjusted for temperature.
- Parameters:
T (float) – Temperature in Kelvin.
comp_idx (int, optional) – Index of compound to calculate property for.
- Returns:
Latent heat of vaporization in J/kg.
- Return type:
np.ndarray
The latent heat of vaporization for each compound at standard pressure and temperature is calculated from the enthalpy of vaporization as:
The heat of vaporization for each compound is then adjusted for variations in temperature[10]:
Liquid molar volume
- fuel.molar_liquid_vol(T, comp_idx=None)
Compute molar liquid volume with temperature correction.
- Parameters:
T (float) – Temperature in Kelvin.
comp_idx (int, optional) – Index of compound to calculate property for.
- Returns:
Molar liquid volume in m^3/mol.
- Return type:
np.ndarray
The liquid molar volume is calculated at a specific temperature \(T\) using the generalized Rackett equation[11] [12] with an updated \(\phi_i\) parameter[10]:
where
Density
- fuel.density(T, comp_idx=None)
Calculate the density of each component at temperature T.
- Parameters:
T (float) – Temperature of the mixture in Kelvin.
comp_idx (int, optional) – Index of compound to calculate property for.
- Returns:
Density of each compound in kg/m^3.
- Return type:
np.ndarray
The density of the i-th compound is given by
Liquid specific heat capacity
- fuel.Cl(T, comp_idx=None)
Compute liquid specific heat capacity in J/kg/K at a given temperature.
- Parameters:
T (float) – Temperature in Kelvin.
comp_idx (int, optional) – Index of compound to calculate property for.
- Returns:
Specific heat capacity in J/kg/K.
- Return type:
np.ndarray
The liquid specific heat capacity for each compound at standard pressure temperature is calculated from the specific heat capacity as:
Saturated vapor pressure
- fuel.psat(T, comp_idx=None, correlation='Lee-Kesler')
Compute saturated vapor pressure.
- Parameters:
T (float) – Temperature in Kelvin.
comp_idx (int, optional) – Index of compound to calculate property for.
correlation (str, optional) – Correlation method (“Ambrose-Walton” or “Lee-Kesler”).
- Returns:
Saturated vapor pressure in Pa.
- Return type:
np.ndarray
The saturated vapor pressure for each compound is calculated as a function of temperature using either the Lee–Kesler method[13] or the Ambrose-Walton method[14]. Both methods solve
for the reduced saturated vapor pressure for each compound, \(p_{r,\text{sat},i} = p_{\text{sat},i}/p_{c,i}\). The default method in FuelLib is the Lee-Kesler method, as it is more stable at higher temperatures. The Lee-Kesler[13] method defines
The Ambrose-Walton[14] correlation sets:
with \(\tau_i = 1 - T_{r,i}\).
- fuel.psat_antoine_coeffs(Tvals=None, units='mks', correlation='Lee-Kesler')
Estimate Antoine coefficients for vapor pressure of an individual compound.
- Parameters:
Tvals (np.ndarray, optional) – Temperature range or nodes for Antoine fit in Kelvin (default [273.15, Tb_i]).
units (str, optional) – Units for pressure in fit (“mks”, “cgs”, “bar”, “atm”)
correlation (str, optional) – Correlation method (“Ambrose-Walton” or “Lee-Kesler”).
- Returns:
Coefficients A, B, C, D
- Return type:
4 np.ndarrays
Users also have the option to return the coefficients from an Antoine fit based on the mixture vapor pressure calculated from Raoult’s law above. Antoine’s equation is:
where \(D_i\) is a conversion factor for converting \(p_{v,i}\) to units of bar (\(D_i = 10^5\)) or dyne/cm 2 (\(D_i = 10^{-1}\)) from Pa. This feature was added to provide Pele users an option for estimating these coefficients for use in CFD simulations with spray. See the PelePhysics documentation for additional information.
Surface tension
- fuel.surface_tension(T, comp_idx=None, correlation='Brock-Bird')
Calculate surface tension of each compound at a given temperature.
- Parameters:
T (float) – Temperature in Kelvin.
comp_idx (int, optional) – Index of compound to calculate property for.
correlation (str, optional) – Correlation method (“Brock-Bird” or “Pitzer”).
- Returns:
Surface tension in N/m.
- Return type:
np.ndarray
Surface tension for each compound is approximated using the relation:
provided \(p_{c,i}\) in bar. The \(Q_i\) term is defined by Brock and Bird[15] (default in FuelLib) as
or by Curl and Pitzer[8] [16] [17] as
Thermal conductivity
- fuel.thermal_conductivity(T, comp_idx=None)
Calculate thermal conductivity at a given temperature.
- Parameters:
T (float) – Temperature in Kelvin.
comp_idx (int, optional) – Index of compound to calculate property for.
- Returns:
Thermal conductivity in W/m/K.
- Return type:
np.ndarray
Thermal conductivity for each compound is computed according to the method of Latini et al. as summarized in Poling’s[8] book:
The constant \(A_i\) is defined by:
provided \(M_{w,i}\) in g/mol. The exponents vary depending on the family of the compound as defined in Thermal conductivity relation parameters. It is assumed that:
aromatics have contain aromatic group contributions (e.g. ACCH)
cycloparaffins contain a ring (e.g. 5-membered ring) and do not contain aromatic groups
olefins contain one or more pairs of carbon atoms linked by a double bond and do not contain aromatic groups or rings
all other compounds are assumed to be saturated hydrocarbons.
Identifier |
Family |
\(A^\ast\) |
\(\alpha\) |
\(\beta\) |
\(\gamma\) |
|---|---|---|---|---|---|
0 |
Saturated hydrocarbons |
0.00350 |
1.2 |
0.5 |
0.167 |
1 |
Aromatics |
0.0346 |
1.2 |
1.0 |
0.167 |
2 |
Cycloparaffins |
0.0310 |
1.2 |
1.0 |
0.167 |
3 |
Olefins |
0.0361 |
1.2 |
1.0 |
0.167 |
Equations for mixture properties from GCM
This section contains correlations for estimating physical properties of the mixture from the individual compound and physical properties defined in Equations for GCM properties and Equations for individual compound correlations. These correlations make it possible to evaluate physical properties at non-standard temperatures and pressures, as the group contribution properties are only defined at standard conditions. The Mixture properties available in FuelLib are listed in table below. Mass and mole fractions defined in Table Mass and mole fractions are used throughout this section.
Symbol |
Units |
Description |
|---|---|---|
\(\rho\) |
kg/m3 |
Density |
\(\nu\) |
m2/s |
Kinematic viscosity |
\(p_v\) |
Pa |
Vapor pressure |
\(\sigma\) |
N/m |
Surface tension |
\(\lambda\) |
W/m/K |
Thermal conductivity |
Symbol |
Definition |
Description |
|---|---|---|
\(Y_i\) |
\(\frac{m_i}{\sum_{k=1}^{N_c} m_k}\) |
Mass fraction of compound i. \(m_i\) is the mass of compound i. |
\(X_i\) |
\(\frac{n_i}{\sum_{k=1}^{N_c} n_k}\) |
Mole fraction of compound i. \(n_i\) is the number of moles compound i. |
Conventional mixing rules
- FuelLib.mixing_rule(var_n, X, pseudo_prop='arithmetic')
Mixing rules for computing mixture properties.
- Parameters:
var_n (np.ndarray) – Individual compound properties.
X (np.ndarray) – Mole fractions of the compounds.
pseudo_prop (str, optional) – Type of mean (“arithmetic” or “geometric”).
- Returns:
Mixture property value.
- Return type:
float
While many of the mixture properties in FuelLib have a unique mixing rule, FuelLib’s mixing_rule function provides a general mixing rule based on the suggestions of Harstad et al[18]. For a given property \(Q\)
where the pseudo-property for the couple of components, \(Q_{ij}\) is computed using an arithmetic,
or a geometric mean,
where \(Q_i\) is the property of the i-th compound of the multicomponent mixture.
Mixture density
- fuel.mixture_density(Yi, T)
Calculate mixture density at a given temperature.
- Parameters:
Yi (np.ndarray) – Mass fractions of each compound.
T (float) – Temperature in Kelvin.
- Returns:
Mixture density in kg/m^3.
- Return type:
float
The mixture’s density is calculated as:
Mixture kinematic viscosity
- fuel.mixture_kinematic_viscosity(Yi, T, correlation='Kendall-Monroe')
Calculate kinematic viscosity of the mixture.
- Parameters:
Yi (np.ndarray) – Mass fractions of each compound.
T (float) – Temperature in Kelvin.
correlation (str, optional) – Mixing model (“Kendall-Monroe” or “Arrhenius”).
- Returns:
Mixture kinematic viscosity in m^2/s.
- Return type:
float
The kinematic viscosity of the mixture is computed using the Kendall-Monroe[19] mixing rule, with an option to use the Arrhenius[20] mixing rule. The viscosity of each component. After evaluating thirty mixing rules, Hernandez et al.[21] found that both Kendall-Monroe and Arrhenius were among the most effective without relying on additional data or parameter fitting. The Kendall-Monroe rule is:
The Arrhenius rule is:
Mixture vapor pressure
- fuel.mixture_vapor_pressure(Yi, T, correlation='Lee-Kesler')
Calculate vapor pressure of the mixture.
- Parameters:
Yi (np.ndarray) – Mass fractions of each compound in the mixture.
T (float) – Temperature in Kelvin.
correlation (str, optional) – Correlation method (“Ambrose-Walton” or “Lee-Kesler”).
- Returns:
Mixture vapor pressure in Pa.
- Return type:
float
The vapor pressure of the mixture is calculated according to Raoult’s law:
- fuel.mixture_vapor_pressure_antoine_coeffs(Yi, Tvals=None, units='mks', correlation='Lee-Kesler')
Estimate Antoine coefficients for vapor pressure of the mixture.
- Parameters:
Yi (np.ndarray) – Mass fractions of each compound in the mixture.
Tvals (np.ndarray, optional) – Temperature range or nodes for Antoine fit in Kelvin (default [273.15, min(Tb)]).
units (str, optional) – Units for pressure in fit (“mks”, “cgs”, “bar”, “atm”)
correlation (str, optional) – Correlation method (“Ambrose-Walton” or “Lee-Kesler”).
- Returns:
Coefficients A, B, C, D
- Return type:
float
Users also have the option to return the coefficients from an Antoine fit based on the mixture vapor pressure calculated from Raoult’s law above. Antoine’s equation is:
where \(D\) is a conversion factor for converting \(p_v\) to units of bar (\(D = 10^5\)) or dyne/cm 2 (\(D = 10^{-1}\)) from Pa. This feature was added to provide Pele users an option for estimating these coefficients for use in CFD simulations with spray. See the PelePhysics documentation for additional information.
Mixture surface tension
- fuel.mixture_surface_tension(Yi, T, correlation='Brock-Bird')
Calculate surface tension of the mixture.
- Parameters:
Yi (np.ndarray) – Mass fractions of each compound in the mixture.
T (float) – Temperature in Kelvin.
correlation (str, optional) – Correlation method (“Pitzer” or “Brock-Bird”).
- Returns:
Mixture surface tension in N/m.
- Return type:
float
The surface tension of the mixture is calculated using the Conventional mixing rules with an arithmetic mean for the pseudo-property \(\sigma_{i,j}\) as recommended by Hugill and van Welsenes[22]:
Mixture thermal conductivity
- fuel.mixture_thermal_conductivity(Yi, T)
Calculate thermal conductivity of the mixture.
- Parameters:
Yi (np.ndarray) – Mass fractions of each compound in the mixture.
T (float) – Temperature in Kelvin.
- Returns:
Thermal conductivity in W/m/K.
- Return type:
float
The thermal conductivity of the mixture is calculated using the power law method of Vredeveld as described in Poling[8]:
Reference Compounds for Jet Fuels
It is difficult to identify individual components of complex multicomponent jet fuels using GCxGC analysis, which generally provides weight percentages of a given hydrocarbon family and carbon number within a sample (e.g., 5% C10 iso-alkane, 2% C13 cycloalkane, etc.). To address this challenge, FuelLib uses a set of reference compounds that represent the major hydrocarbon families and carbon numbers found in jet fuels. A comprehensive list of the reference compounds used in FuelLib can be found in the fuelData/refCompounds.csv file, with associated functional group decompositions in fuelData/groupDecompositionData/refCompounds.csv.
For ease of reference, the reference compounds and keys corresponding to a PelePhysics mechanism fuellib_posf_nonreacting are provided in the table below.
When provided, the PelePhysics keys can be used to link the compounds in FuelLib to species in PelePhysics simulations via Export4Pele.py as described in Exporting to PelePhysics.
GCxGC Bin |
Formula |
Reference Compound |
PelePhysics Key |
|---|---|---|---|
Toluene |
C7H8 |
toluene |
C6H5CH3 |
C2-Benzene |
C8H10 |
ethyl benzene |
C6H5C2H5 |
C3-Benzene |
C9H12 |
propyl benzene |
C6H5C3H7 |
C4-Benzene |
C10H14 |
butyl benzene |
C6H5C4H9 |
C5-Benzene |
C11H16 |
pentyl benzene |
C6H5C5H11 |
C6-Benzene |
C12H18 |
hexyl benzene |
C6H5C6H13 |
C7-Benzene |
C13H20 |
heptyl benzene |
C6H5C7H15 |
C8-Benzene |
C14H22 |
octyl benzene |
C6H5C8H17 |
C9-Benzene |
C15H24 |
nonyl benzene |
C6H5C9H19 |
C10-Benzene |
C16H26 |
decyl benzene |
C6H5C10H21 |
Diaromatic-C10 |
C10H8 |
naphthalene |
NAPH |
Diaromatic-C11 |
C11H10 |
1-methyl naphthalene |
METHYLNAPH-1 |
Diaromatic-C12 |
C12H12 |
1-ethyl naphthalene |
ETHYLNAPH-1 |
Diaromatic-C13 |
C13H14 |
1-propyl naphthalene |
PROPYLNAPH-1 |
Diaromatic-C14 |
C14H16 |
1-butyl naphthalene |
BUTYLNAPH-1 |
Cycloaromatic-C09 |
C9H10 |
indane |
INDANE |
Cycloaromatic-C10 |
C10H12 |
tetralin |
TETRA |
Cycloaromatic-C11 |
C11H14 |
2-methyl tetralin |
METHYLTETRA-2 |
Cycloaromatic-C12 |
C12H16 |
2-ethly tetralin |
ETHYLTETRA-2 |
Cycloaromatic-C13 |
C13H18 |
2-propyl tetralin |
PROPYLTETRA-2 |
Cycloaromatic-C14 |
C14H20 |
2-butyl tetralin |
BUTYLTETRA-2 |
Cycloaromatic-C15 |
C15H22 |
2-pentyl tetralin |
PENTYLTETRA-2 |
C07-Isoparaffin |
C7H16 |
2-methyl hexane |
C7H16-2 |
C08-Isoparaffin |
C8H18 |
2-methyl heptane |
C8H18-2 |
C09-Isoparaffin |
C9H20 |
2-methyl octane |
C9H20-2 |
C10-Isoparaffin |
C10H22 |
2-methyl nonane |
C10H22-2 |
C11-Isoparaffin |
C11H24 |
2-methyl decane |
C11H24-2 |
C12-Isoparaffin |
C12H26 |
2-methyl undecane |
C12H26-2 |
C13-Isoparaffin |
C13H28 |
2-methyl dodecane |
C13H28-2 |
C14-Isoparaffin |
C14H30 |
2-methyl tridecane |
C14H30-2 |
C15-Isoparaffin |
C15H32 |
2-methyl tetradecane |
C15H32-2 |
C16-Isoparaffin |
C16H34 |
2-methyl pentadecane |
C16H34-2 |
C17-Isoparaffin |
C17H36 |
2-methyl hexadecane |
C17H36-2 |
C18-Isoparaffin |
C18H38 |
2-methyl heptadecane |
C18H38-2 |
C19-Isoparaffin |
C19H40 |
2-methyl octadecane |
C19H40-2 |
C20-Isoparaffin |
C20H42 |
2-methyl nonadecane |
C20H42-2 |
C21-Isoparaffin |
C21H44 |
2-methyl icosane |
C21H44-2 |
C22-Isoparaffin |
C22H46 |
2-methyl henicosane |
C22H46-2 |
C23-Isoparaffin |
C23H48 |
2-methyl docosane |
C23H48-2 |
C24-Isoparaffin |
C24H50 |
2-methyl tricosane |
C24H50-2 |
n-C07 |
C7H16 |
n-heptane |
NC7H16 |
n-C08 |
C8H18 |
n-octane |
NC8H18 |
n-C09 |
C9H20 |
n-nonane |
NC9H20 |
n-C10 |
C10H22 |
n-decane |
NC10H22 |
n-C11 |
C11H24 |
n-undecane |
NC11H24 |
n-C12 |
C12H26 |
n-dodecane |
NC12H26 |
n-C13 |
C13H28 |
n-tridecane |
NC13H28 |
n-C14 |
C14H30 |
n-tetradecane |
NC14H30 |
n-C15 |
C15H32 |
n-pentadecane |
NC15H32 |
n-C16 |
C16H34 |
n-hexadecane |
NC16H34 |
n-C17 |
C17H36 |
n-heptadecane |
NC17H36 |
n-C18 |
C18H38 |
n-octadecane |
NC18H38 |
n-C19 |
C19H40 |
n-nonadecane |
NC19H40 |
n-C20 |
C20H42 |
n-icosane |
NC20H42 |
n-C21 |
C21H44 |
n-henicosane |
NC21H44 |
n-C22 |
C22H46 |
n-docosane |
NC22H46 |
n-C23 |
C23H48 |
n-tricosane |
NC23H48 |
C07-Monocycloparaffin |
C7C14 |
methyl cyclohexane |
C6H11CH3 |
C08-Monocycloparaffin |
C8H16 |
ethyl cyclohexane |
C6H11C2H5 |
C09-Monocycloparaffin |
C9H18 |
propyl cyclohexane |
C6H11C3H7 |
C10-Monocycloparaffin |
C10H20 |
butyl cyclohexane |
C6H11C4H9 |
C11-Monocycloparaffin |
C11H22 |
pentyl cyclohexane |
C6H11C5H11 |
C12-Monocycloparaffin |
C12H24 |
hexyl cyclohexane |
C6H11C6H13 |
C13-Monocycloparaffin |
C13H26 |
heptyl cyclohexane |
C6H11C7H15 |
C14-Monocycloparaffin |
C14H28 |
octyl cyclohexane |
C6H11C8H17 |
C15-Monocycloparaffin |
C15H30 |
nonyl cyclohexane |
C6H11C9H19 |
C16-Monocycloparaffin |
C16H32 |
decyl cyclohexane |
C6H11C10H21 |
C17-Monocycloparaffin |
C17H34 |
undecyl cyclohexane |
C6H11C11H23 |
C18-Monocycloparaffin |
C18H36 |
dodecyl cyclohexane |
C6H11C12H25 |
C19-Monocycloparaffin |
C19H38 |
tridecyl cyclohexane |
C6H11C13H27 |
C08-Dicycloparaffin |
C8H14 |
Octahydropentalene |
OHPEN |
C09-Dicycloparaffin |
C9H16 |
Hydrindane |
HYDRIND |
C10-Dicycloparaffin |
C10H18 |
Decalin |
DECALIN |
C11-Dicycloparaffin |
C11H20 |
2-methyldecalin |
METHYLDECALIN-2 |
C12-Dicycloparaffin |
C12H22 |
2-ethyldecalin |
ETHYLDECALIN-2 |
C13-Dicycloparaffin |
C13H24 |
2-propyldecalin |
PROPYLDECALIN-2 |
C14-Dicycloparaffin |
C14H26 |
2-butyldecalin |
BUTYLDECALIN-2 |
C15-Dicycloparaffin |
C15H28 |
2-pentyldecalin |
PENTYLDECALIN-2 |
C16-Dicycloparaffin |
C16H30 |
2-hexyldecalin |
HEXYLDECALIN-2 |
C17-Dicycloparaffin |
C17H32 |
2-heptyldecalin |
HEPTYLDECALIN-2 |
C10-Tricycloparaffin |
C10H16 |
C1CC2C(C1)C1CCCC21 |
TRICYCLO-C10 |
C11-Tricycloparaffin |
C11H18 |
C1CC2CC3CCCC3C2C1 |
TRICYCLO-C11 |
C12-Tricycloparaffin |
C12H20 |
C1CC2CC3CCCC3CC2C1 |
TRICYCLO-C12 |
C13-Tricycloparaffin |
C13H22 |
C1CCC2C(C1)CC1CCCCC12 |
TRICYCLO-C13 |
C14-Tricycloparaffin |
C13H22 |
C1CCC2CC3CCCCC3CC2C1 |
TRICYCLO-C14 |
C10-Alkene |
C10H20 |
1-decene |
DECENE-1 |
C12-Alkene |
C12H24 |
1-dodecene |
DODECENE-1 |
C14-Alkene |
C14H28 |
1-tetradecene |
TETRADECENE-1 |
C16-Alkene |
C16H32 |
1-hexadecene |
HEXADECENE-1 |
Validation
Single Component Fuels
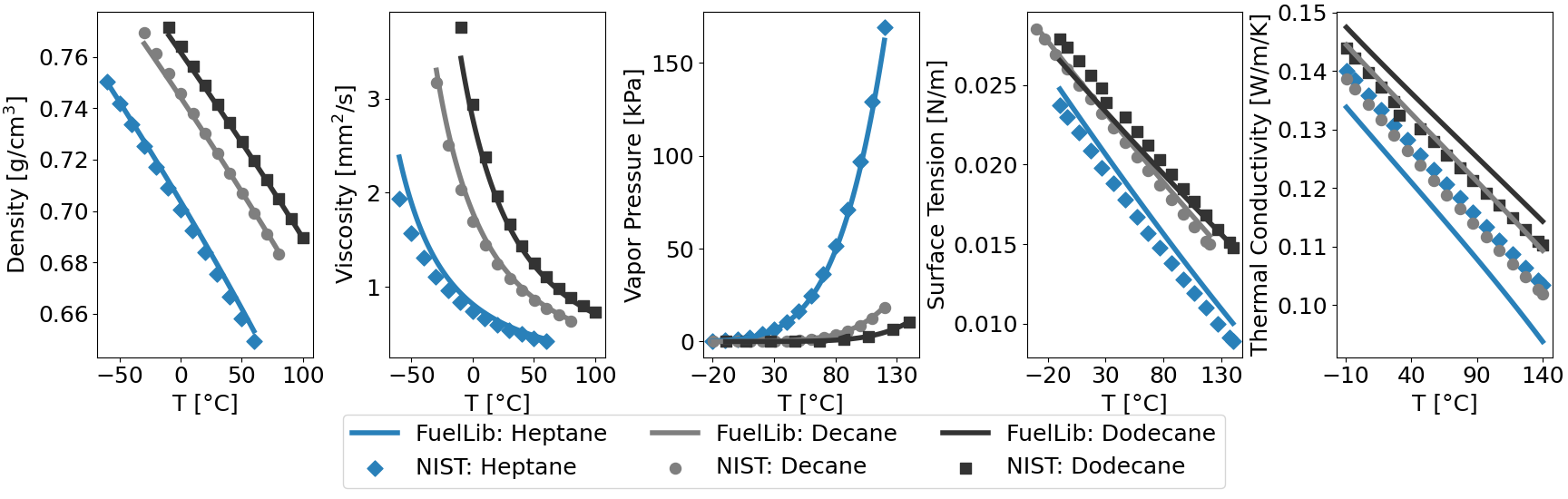
Fig. 1 Properties of heptane, decane, and dodecane against predictive data from NIST Chemistry WebBook.
Multicomponent Fuels
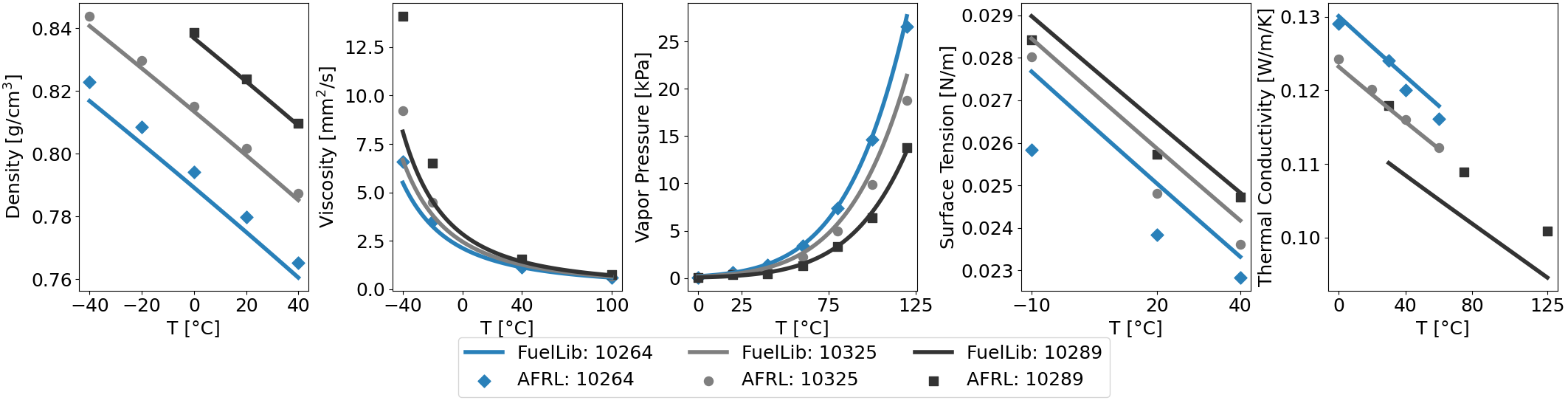
Fig. 2 Properties of conventional jet fuels JP-8 (POSF10264), Jet A (POSF10325), and JP-5 (POSF10289) against data from the Air Force Research Laboratory[5]. Note that the data sets for thermal conductivity are very inconsistent, but they typically show linear decreases in thermal conductivity with temperature.
Fuel Blends
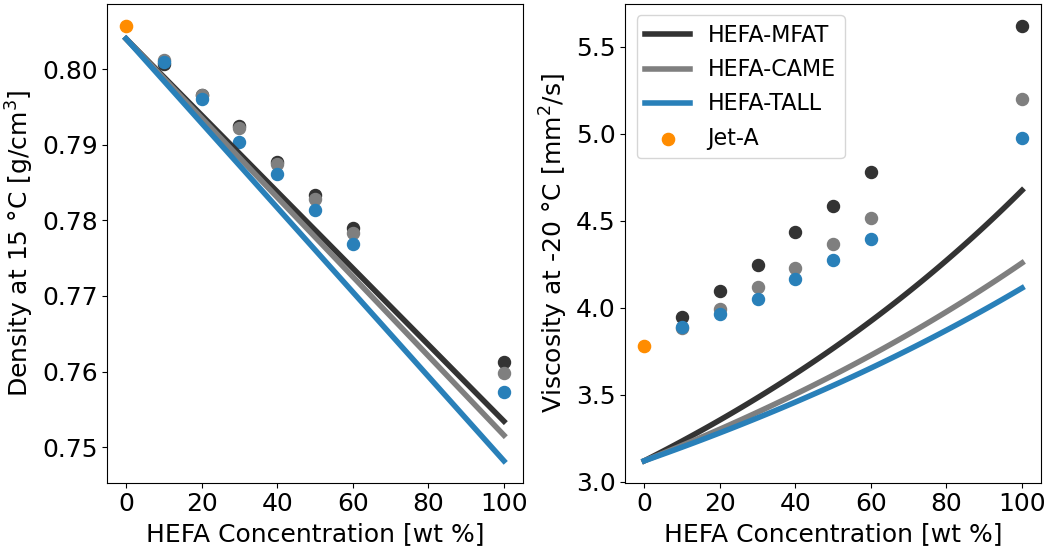
Fig. 3 Properties of three HEFA fuels produced from different feedstocks (camelina, tallow, and mixed fat) blended with Jet-A. Measurement and GCxGC data from Vozka et al.[6].